SaaS
“LFS-HIT Screening” quickly and more precisely screens novel HIT candidates through AI-based numerous calculations in a premium library, with or without reference structure data. Plus, our technical cooperation has reduced the time from synthesis to customs clearance and receipt by half again. A proven platform in various therapeutic areas and protein families, including kinase, GPCR, and nuclear receptors. Discover novel scaffolds and HIT candidates from a library of up to 50 million premium compounds.
Are you planning to develop NME? Want to discover a novel scaffold HIT?
Start your planning with the premium library of up to 50 million capable of screening novel scaffolds for NME development. Strengthen your rights by securing material patents from the HIT discovery stage.
Want to find HITs that have a clear binding mode from the start?
STB offers HIT candidates with a clear binding mode as best-pose prediction between target and compound.
Do you have trouble with the long wait for the synthesis or customs clearance?
Save synthesis time/cost for verification with HIT candidates that can be synthesized quickly and reliably.

ALFS Library (Large-scale and Fast-synthesis DB)
Input : Target protein info. Ref. drug info. (if available)
BDeep Learning Screening
Input : 50M Cpds list & Target 3D structures
Output : Top ranked 500 Cpds & poses info.
CFalse Positives Filtering
Input : Poses of the top ranked 500 Cpds
Output : List of 200 Cpds with false positives filtered
DHIT Selection
Input : The screening score of 200 Cpds
Output : Priority of ~200 Cpds list
Auto-Hit Discovery by DeepMatcher®-Hit
Flow diagram of Hit discovery by DeepMatcher® Compounds are prescreened through three steps before being docked in a pocket of the target. Once the optimal pose is selected, the MD simulation is finally performed in a total of five steps.
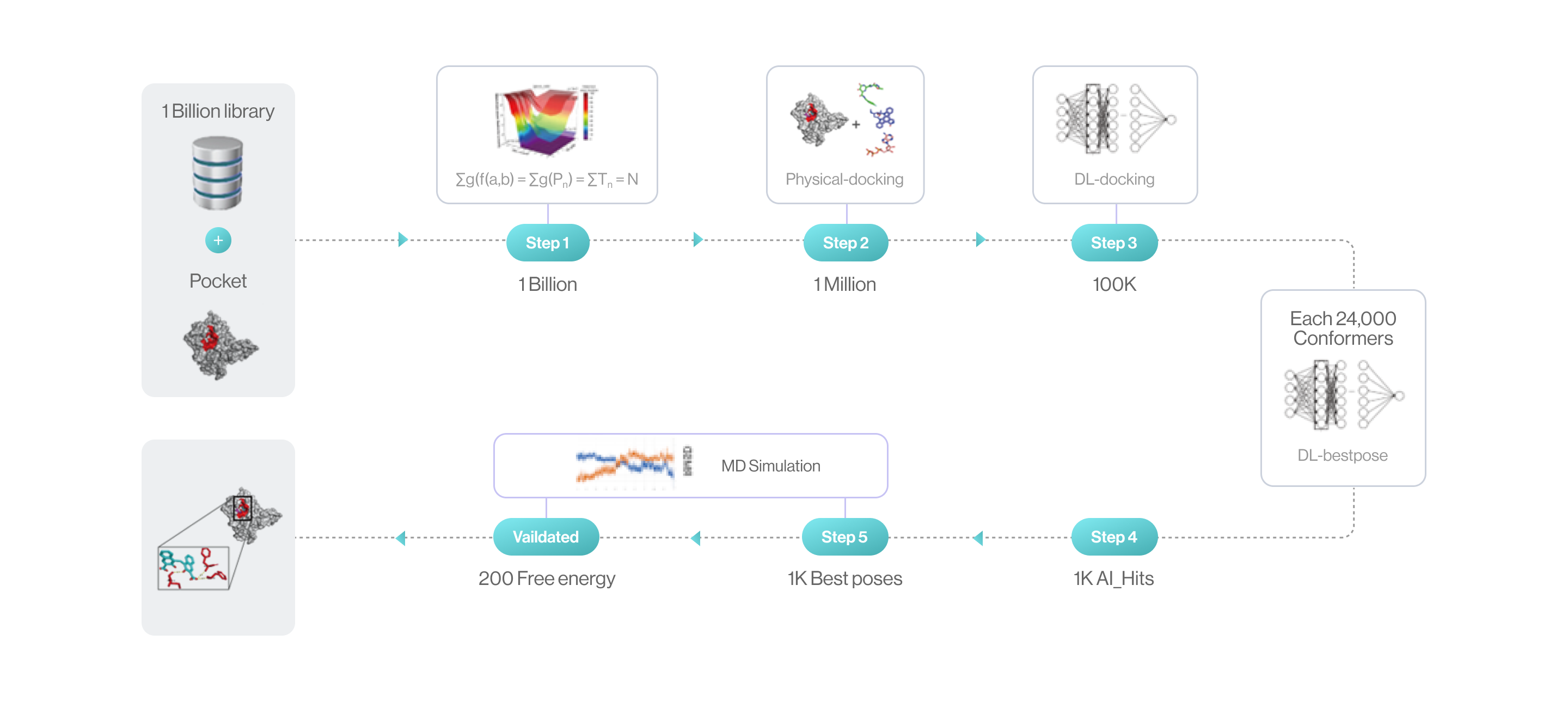
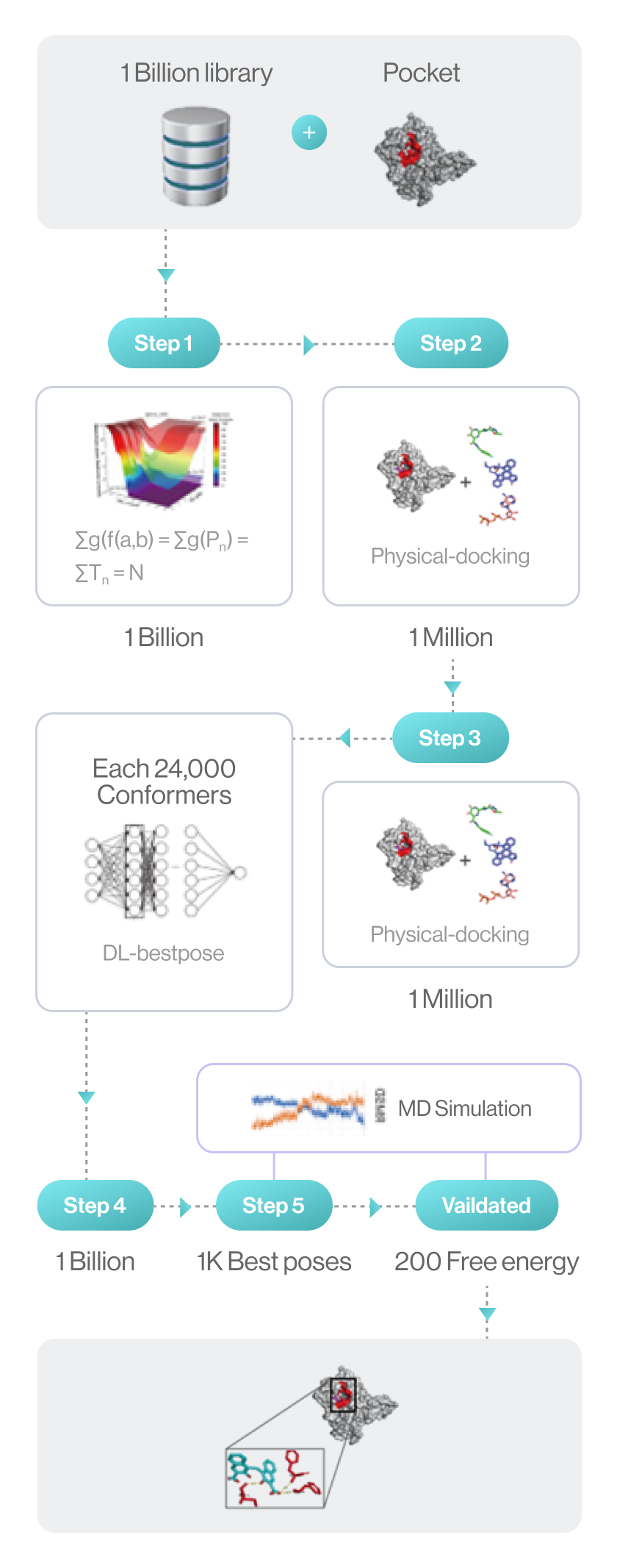
Syntekabio launched STB CLOUD in late 2022, a suite of cloud-based proprietary platforms, to meet the unmet needs of these cost-conscious innovators seeking reliable yet affordable new drug candidate discovery services. STB CLOUD screens 1 billion purchasable compounds based on unique featurization method for 3D information-based data and 1 million potential candidate compounds
One million chemicals selected from a library of 1 billion compounds was docked into target 3D structure using command line interface (CLI) based in-house structure manipulation package.
Figure2. Chemicallibrary
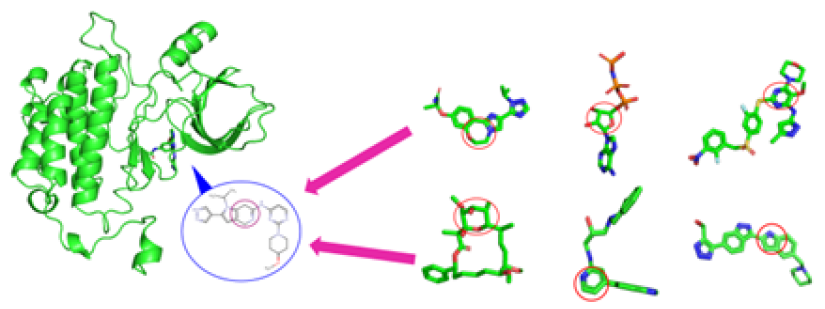
Figure3. Methods for centroidal atom vector-based fast protein-ligand docking. After docking, three atom-vector based rotations along the core ring. It rotates while moving 1000 times in each revolution and then, generates a protein-ligand interaction (PLI) score
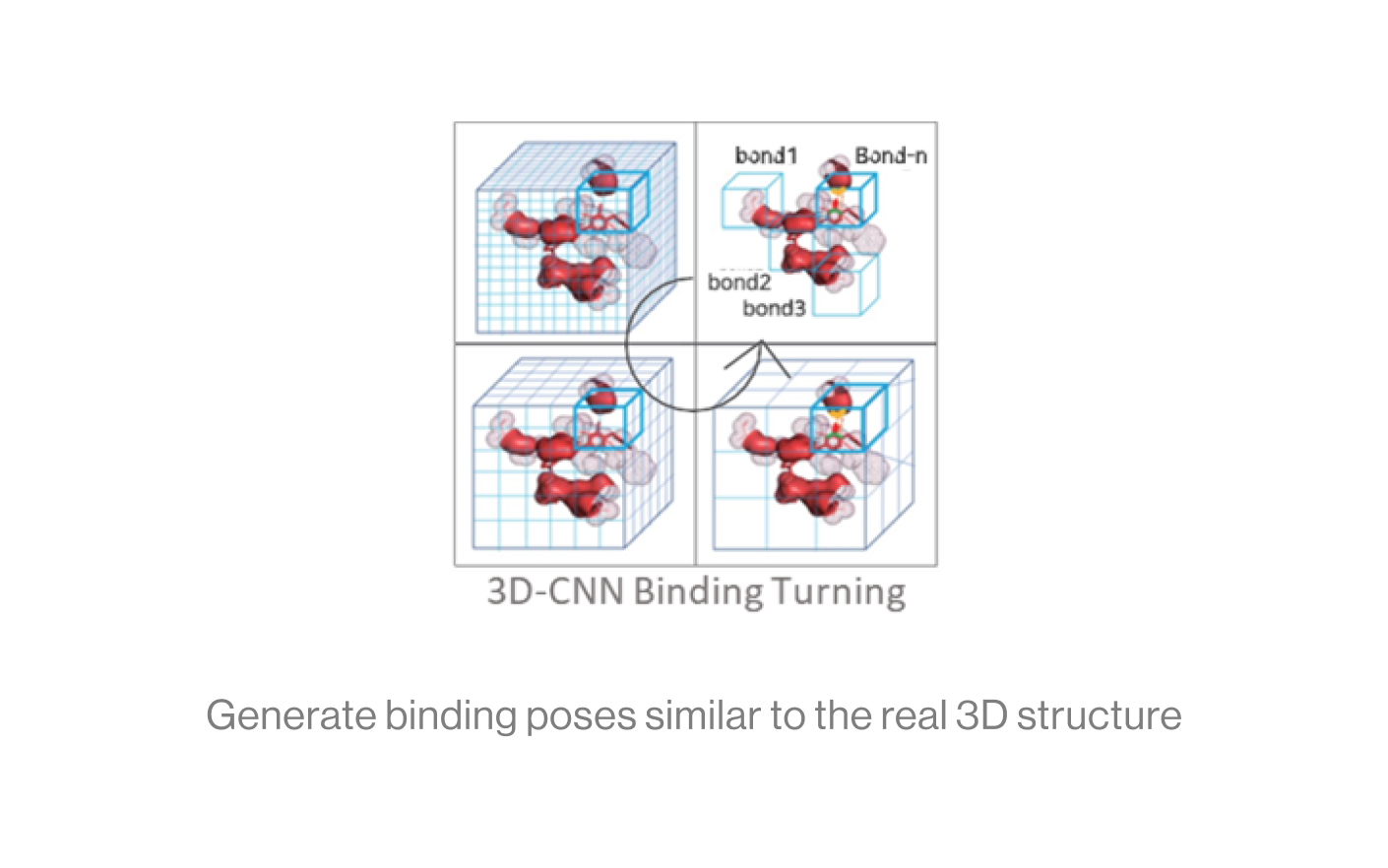
The diagram below shows the interaction between a protein and a ligand in the pocket as a grid box, a 3D-CNN based intermediate stage of repeated max pooling and 3D convolution, generating feature maps, and representing bonds. Here, the starting grid box (48) and kernel size (3*3*3) are used. The density function is based on the geometry of adjacent bonds of two atoms. Therefore, the sum of bonds is defined as binding affinity.
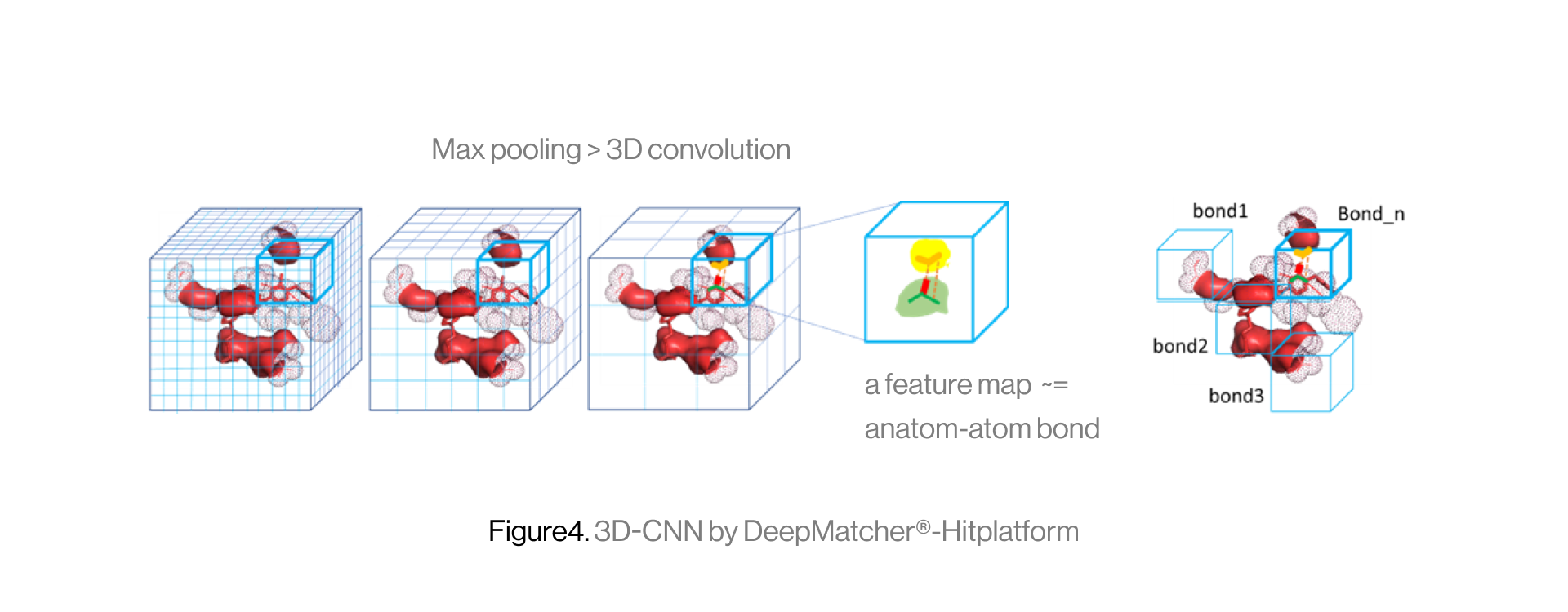
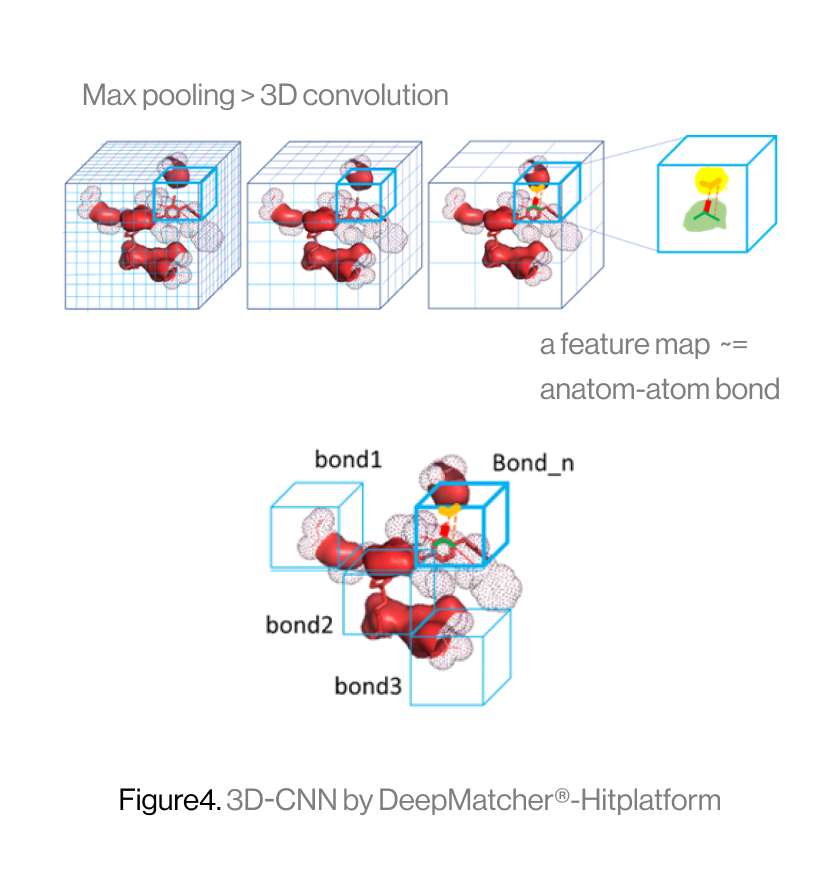
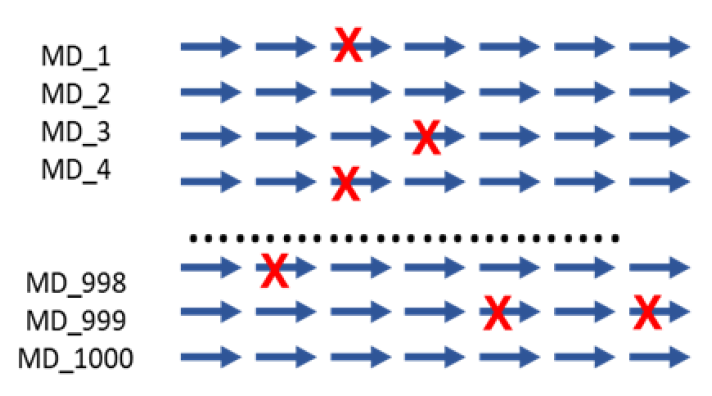
Figure5. Auto-MD simulation
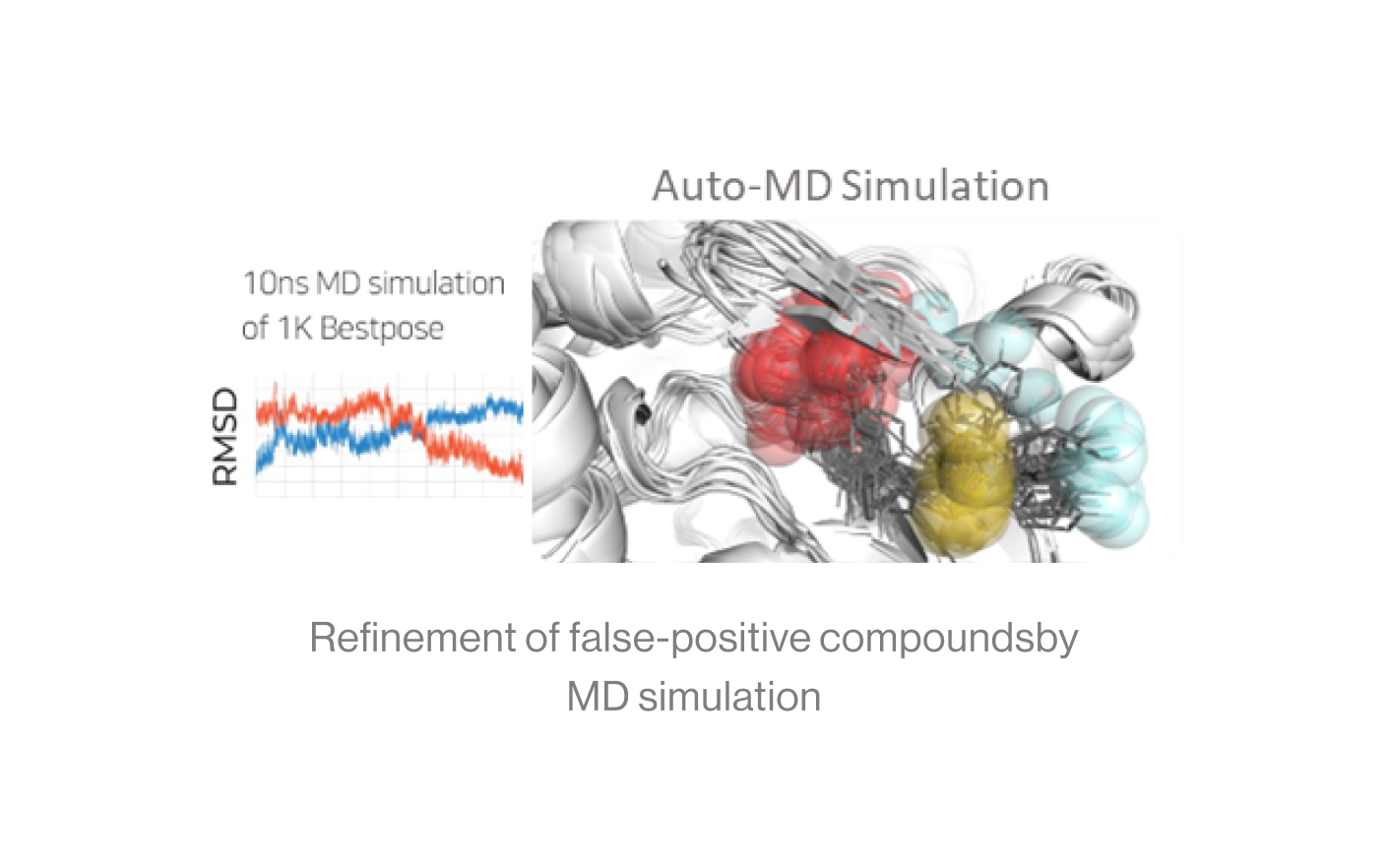
Auto-MD Simulation
Syntekabio's fully automated MD simulation algorithm performs molecular dynamics simulations (10 ns) in explicit water box for 1,000 protein-ligand complexes.
The diagram shows an example of running continuously from a dead step during 1,000 MD simulations. If one runs 1000 MD simulations at once, a large number of simulations may crash and die in certain cases. However, in the automated environment, it proceeds by saving the checkpoint and resume from the saved point.
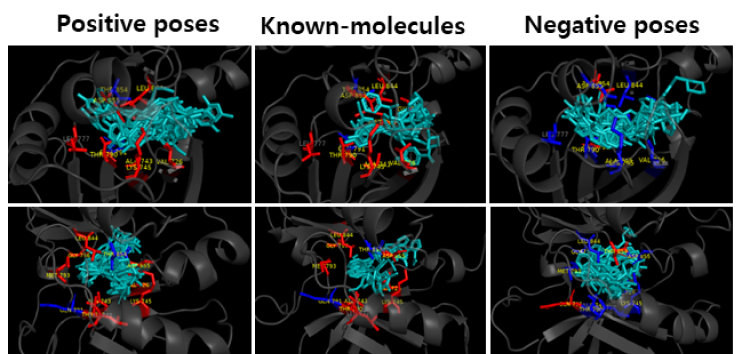
Figure 6. Comparison of AI-hit positive andnegative
predictions with known Molecules.
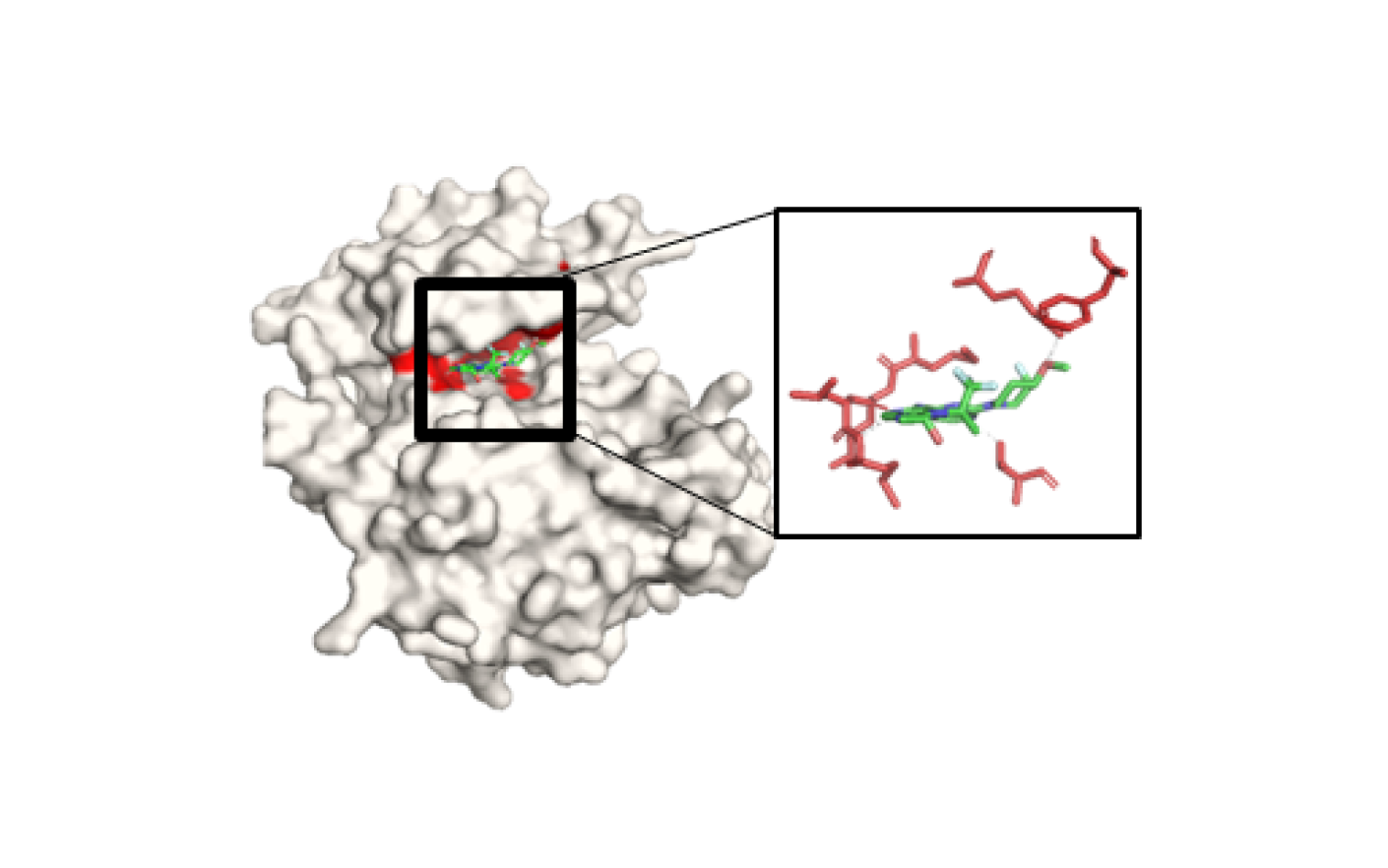
Key-Residue Analysis
The known molecules and the predicted molecules with EGFR protein were compared to see if the functional (Hinge, DFG) and conformational keys (G-loop, M766 in c-Helix) were well matched. In the case of positive prediction, the functional and conformational keys were well matched, whereas in case of negative, there were few matched residues. (Red: strong binding, Blue: weak binding)
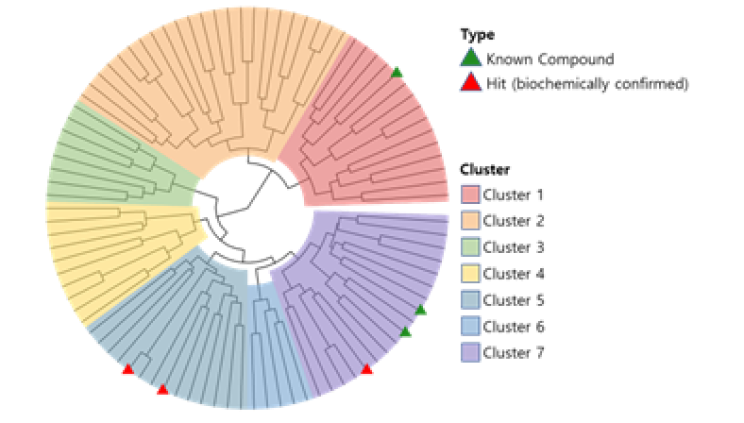
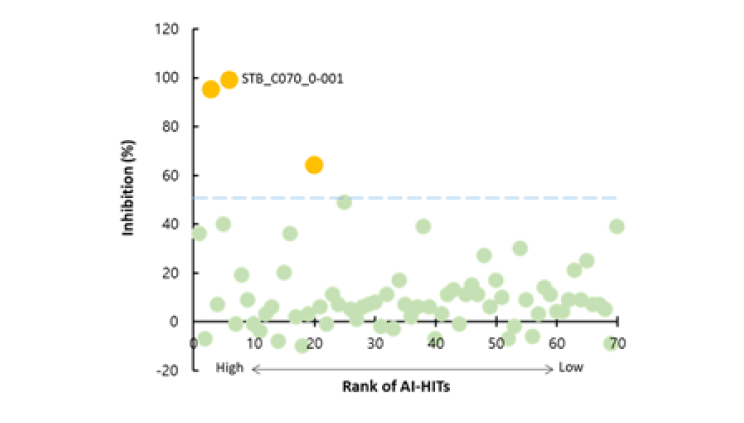
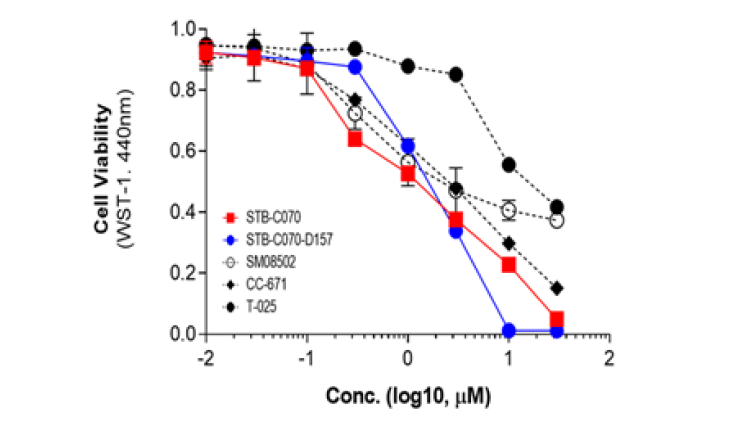
CLK2, also known as CDC-like kinase 2, is a protein kinase that plays a role in the regulation of pre-mRNA splicing. Presented 89 candidate compounds using Deepmatcher®-Hit as a result of structure analysis using tanimoto coefficient with KNN algorithm. a) It was confirmed that 89 compounds were divided into 7 groups. b) Purchasable compounds were biochemically confirmed their inhibition activities. Three compounds of 74 purchased candidates (yellow circles) inhibited enzyme activity of CLK2 more than 50%. c) The growth inhibitory effect of each cancer cell by treatment with STB-C070, STB-C070-D157 and reference compounds were analyzed using WST-1 assay. GI50 was calculated from the 8-point dose-response curves by triplicate test.
a) DeepMatcher® Result analysis
b) Biochemical assay result of AI-hits
c) Cell-based assay result of AI-hits
Expo-ro 1, Expo Tower #1903,
Yuseong-gu, Daejeon,
Republic of Korea
Headquarter
Samunan-ro 92, Gwanghwamun Officia Building #1708,
Jongno-gu, Seoul, Republic of Korea
Business Center
425 Fifth Avenue Suite 505 New York,
NY, USA #10016
Syntekabio USA Inc.
![]()
© 2024 Syntekabio, Inc. All rights reserved.


